
Contact information
Email: david.stellwagen [at] mcgill.ca
Tel.: 514-934-1934 ext. 42806
Academic affiliations
Associate Professor | Neurology & Neurosurgery, Medicine (Dept. & Faculty)
Researcher | Research Institute of the McGill University Health Centre
Lab Website
Publications
Research
TNFα Regulation of Synaptic Plasticity and Neuronal Function
We are studying the role of the pro-inflammatory cytokine Tumor Necrosis Factor-alpha (TNFα), a molecule principally characterized in the immune system, in the regulation of synaptic transmission. TNFα has an intrinsic neuronal function as it, through the alteration of receptor trafficking, acts as a glia-released mediator of homeostatic synaptic scaling, an important form of synaptic plasticity. Further, TNFα regulates the surface expression of calcium-permeable glutamate receptors, which will greatly increase a neuron's vulnerability to excitotoxicity, one of the major causes of cell death following neural trauma. This is opening up a new area of research for understanding both normal neuronal function and neuropathology. We are interested in characterizing the role of TNFα and other cytokines in the normally functioning nervous system, to understand how the nervous system will be damaged by excessive release of these same cytokines during neuroinflammation.
Defining the function of TNFα in the synaptic plasticity

Synaptic plasticity is one of the fundamental characteristics of the nervous systems, and is thought to underlie learning and memory, including aberrant forms of learning such as drug addiction and neuropathic pain, as well as the activity-dependent refinement of connectivity observed during development. Synaptic plasticity is generally believed to be a result, at least in part, from the alteration in the number of AMPA-type glutamate receptors found at excitatory synapses. Recently, we have identified TNFα as a novel regulator of AMPA receptor trafficking, resulting in a dramatic and rapid exocytosis of AMPA receptors. Furthermore, TNFα is a glia-released factor, whose continual release is maintaining the surface expression of a population of AMPARs. TNFα also results in the insertion of a particular subtype of AMPAR, the GluR2-lacking AMPAR, which is Ca2+ permeable and therefore could also alter the threshold for synaptic plasticity or excitotoxicity. This subclass of receptor has also been implicated in a number of disease states. Simultaneously, TNFα also regulates neuronal inhibition, by causing an endocytosis of the GABA-A receptor, the principle mediator of fast inhibition in the brain. The receptor trafficking induced by TNFα underlies homeostatic synaptic plasticity.
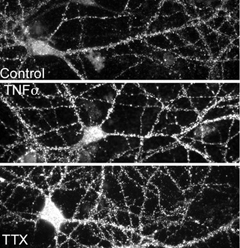
Homeostatic synaptic plasticity entails the uniform adjustments in the strength of all synapses on a given cell in response to prolonged changes in the cell’s electrical activity, and is critical for the stability of neuronal circuits. In the case of prolonged period of low activity, the excitatory synapses on primary neurons are strengthened due to insertion of additional AMPA receptors while the inhibitory synapses weaken, due to removal of GABA receptors. TNFα is a critical mediator of these changes. Interfering with TNFα signaling, either pharmacologically or genetically, prevents the changes in synaptic strength induced by chronic activity blockade. Moreover, the TNFα is of glial origin, suggesting glia detect neuronal activity and feedback homeostatic signals, including TNFα. We can now probe the role of TNFα and homeostatic plasticity in the nervous system. Removing TNFα signaling interferes with the ocular dominance plasticity observed in the visual cortex following monocular deprivation, suggesting a role for homeostatic plasticity in this process. We are interested in exploring other role for this form of plasticity in nervous system function.
Defining the function of TNFα in disease and trauma


The lab is focusing on three main questions:
1) The detailed mechanisms of glial-neuronal communication and TNFα-dependent synaptic plasticity
2) Using the TNFα knockout mouse to investigate the role of TNFα-dependent plasticity in development and behavior
3) Investigate the role of TNFα-dependent receptor trafficking (particularly of the calcium permeable AMPA receptor) to neuro-inflammatory insults